









Konten
Sklerosis tuberous Bourneville
Apa itu ?
Bourneville tuberous sclerosis adalah penyakit genetik kompleks yang ditandai dengan perkembangan tumor jinak (non-kanker) di berbagai bagian tubuh. Tumor ini kemudian dapat ditemukan di kulit, otak, ginjal, dan organ serta jaringan lain. Patologi ini juga dapat menyebabkan masalah serius dalam perkembangan individu. Namun, manifestasi klinis dan tingkat keparahan penyakit bervariasi dari pasien ke pasien.
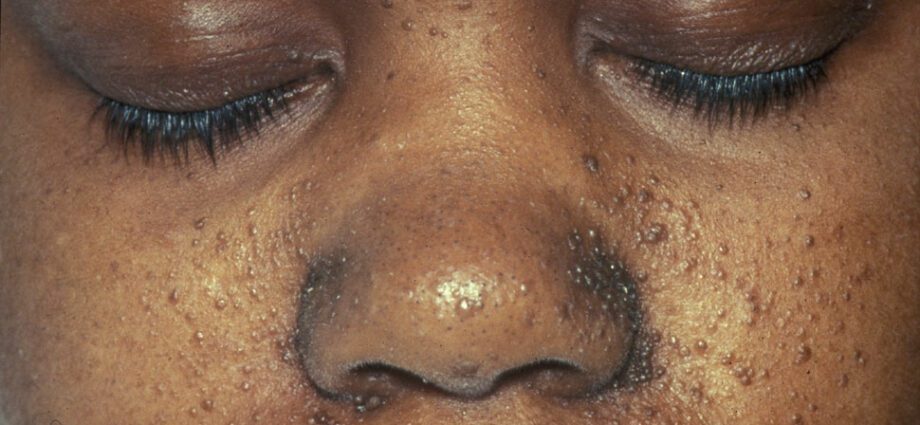
Kelainan kulit yang terkait umumnya mirip dengan bintik-bintik pada kulit atau area di mana kulit lebih terang daripada bagian tubuh lainnya. Perkembangan tumor di wajah disebut angiofibroma.
Dalam konteks kerusakan otak, tanda-tanda klinisnya adalah serangan epilepsi, masalah perilaku (hiperaktivitas, agresivitas, cacat intelektual, masalah belajar, dll.). Beberapa anak dengan penyakit ini bahkan memiliki beberapa bentuk autisme, gangguan perkembangan, yang mempengaruhi interaksi sosial dan komunikasi. Tumor otak jinak juga bisa menyebabkan komplikasi yang bisa berakibat fatal bagi penderitanya.
Perkembangan tumor di ginjal umum terjadi pada orang dengan tuberous sclerosis. Hal ini dapat menyebabkan komplikasi parah pada fungsi ginjal. Selain itu, tumor dapat berkembang di jantung, paru-paru dan retina. (2)
Ini adalah penyakit langka, yang prevalensinya (jumlah kasus pada populasi tertentu pada waktu tertentu) berjumlah 1 / 8 hingga 000 / 1 orang. (15)
Gejala
Manifestasi klinis yang terkait dengan tuberous sclerosis dari Bourneville bervariasi sesuai dengan organ yang terkena. Selain itu, gejala yang terkait dengan penyakit ini sangat bervariasi dari satu orang ke orang lain. Dengan gejala mulai dari ringan hingga berat.
Gejala yang paling banyak diidentifikasi dari penyakit ini termasuk serangan epilepsi, gangguan kognitif dan perilaku, kelainan kulit, dll. Organ yang paling sering terkena adalah: otak, jantung, ginjal, paru-paru dan kulit.
Perkembangan tumor ganas (kanker) mungkin terjadi pada penyakit ini tetapi jarang terjadi dan terutama mempengaruhi ginjal.
Tanda-tanda klinis penyakit di otak berasal dari serangan pada tingkat yang berbeda:
– kerusakan pada tuberkel kortikal;
– nodul ependimal (SEN);
- astrositoma ependymal raksasa.
Mereka menghasilkan: perkembangan keterbelakangan mental, kesulitan belajar, gangguan perilaku, agresivitas, gangguan perhatian, hiperaktif, gangguan obsesif-kompulsif, dll.
Kerusakan ginjal ditandai dengan perkembangan kista atau angiomyolipoma. Ini dapat menyebabkan sakit ginjal dan bahkan gagal ginjal. Jika perdarahan berat terlihat, mungkin karena anemia berat atau tekanan darah tinggi. Konsekuensi lain yang lebih serius tetapi jarang juga dapat terlihat, khususnya perkembangan karsinoma (tumor sel penyusun epitel).
Kerusakan mata bisa mirip dengan bintik-bintik yang terlihat di retina, menyebabkan gangguan penglihatan atau bahkan kebutaan.
Kelainan kulit sangat banyak:
– makula hipomelanik: yang mengakibatkan munculnya bintik-bintik terang pada kulit, di bagian tubuh mana pun, sebagai akibat dari kekurangan melanin, suatu protein yang memberi warna pada kulit;
– munculnya bintik-bintik merah di wajah;
- bercak berubah warna di dahi;
– kelainan kulit lainnya, tergantung dari satu individu ke individu lainnya.
Lesi paru terjadi pada 1/3 pasien dengan sedikit dominasi wanita. Gejala yang terkait kemudian lebih atau kurang parah kesulitan bernapas.
Asal usul penyakit
Asal usul penyakit ini adalah genetik dan keturunan.
Transmisi melibatkan mutasi pada gen TSC1 dan TSC2. Gen-gen yang menarik ini berperan dalam pembentukan protein: hamartin dan tuberin. Kedua protein ini memungkinkan, melalui permainan interaktif, untuk mengatur proliferasi sel.
Pasien dengan penyakit ini dilahirkan dengan setidaknya satu salinan mutasi gen ini di setiap sel mereka. Mutasi ini kemudian membatasi pembentukan hamartine atau tubertine.
Dalam konteks di mana dua salinan gen bermutasi, mereka sepenuhnya mencegah produksi kedua protein ini. Oleh karena itu, kekurangan protein ini tidak lagi memungkinkan tubuh untuk mengatur pertumbuhan sel-sel tertentu dan, dalam pengertian ini, mengarah pada perkembangan sel tumor di berbagai jaringan dan/atau organ.
Faktor risiko
Faktor risiko untuk mengembangkan patologi semacam itu adalah genetik.
Memang, penularan penyakit ini efektif melalui mode autosomal dominan. Entah, gen minat yang bermutasi terletak di kromosom non-seksual. Selain itu, kehadiran hanya satu dari dua salinan gen yang bermutasi sudah cukup untuk mengembangkan penyakit.
Dalam pengertian ini, seorang individu yang memiliki salah satu dari dua orang tua yang menderita penyakit ini memiliki risiko 50% mengembangkan fenotipe yang sakit itu sendiri.
Pencegahan dan perawatan
Diagnosis penyakit ini pertama-tama diferensial. Hal ini didasarkan pada kriteria fisik atipikal. Dalam kebanyakan kasus, tanda-tanda karakteristik pertama dari penyakit ini adalah: adanya serangan epilepsi berulang dan keterlambatan perkembangan subjek. Dalam kasus lain, tanda-tanda pertama ini menghasilkan bintik-bintik kulit atau identifikasi tumor jantung.
Setelah diagnosis pertama ini, pemeriksaan tambahan sangat penting untuk memvalidasi diagnosis atau tidak. Ini termasuk:
– pemindaian otak;
– MRI (Magnetic Resonance Imaging) otak;
- USG jantung, hati dan ginjal.
Diagnosis dapat efektif pada saat kelahiran anak. Jika tidak, penting untuk dilakukan secepat mungkin untuk menangani pasien sesegera mungkin.
Saat ini, tidak ada obat untuk penyakit tersebut. Oleh karena itu, perawatan terkait tidak tergantung pada gejala yang disajikan oleh masing-masing individu.
Biasanya, obat anti epilepsi diberikan untuk membatasi kejang. Selain itu, obat untuk pengobatan sel tumor otak dan ginjal juga diresepkan. Dalam konteks masalah perilaku, perawatan khusus anak diperlukan.
Pengobatan penyakit ini biasanya jangka panjang. (1)